論文
当研究室から発表した論文を、最新のものから順にリストしています。当研究室のメンバーとして論文に貢献した著者を太字で示しています。当研究室のメンバーが筆頭あるいは責任著者を務める論文にはプレビュー画像を付しています。†: 共筆頭著者. *: (共)責任著者.
2026
-
 Species Spotlight Australian pitcher plant (Cephalotus follicularis)Nature Ecology & Evolution : . Feb 2026
Species Spotlight Australian pitcher plant (Cephalotus follicularis)Nature Ecology & Evolution : . Feb 2026 -
 News & Views Genomic cradle for thousandsNature Plants 12: 271-272. Jan 2026
News & Views Genomic cradle for thousandsNature Plants 12: 271-272. Jan 2026 -
 Review Decoding plant cell heterogeneity and dynamics across responses, development, to evolution with single-cell technologiesCurrent Opinion in Plant Biology 90: 102854. Apr 2026
Review Decoding plant cell heterogeneity and dynamics across responses, development, to evolution with single-cell technologiesCurrent Opinion in Plant Biology 90: 102854. Apr 2026 -
 Original Transcriptomic prey-capture responses in convergently evolved carnivorous pitcher plantsNew Phytologist 249: 2559-2573. Feb 2026
Original Transcriptomic prey-capture responses in convergently evolved carnivorous pitcher plantsNew Phytologist 249: 2559-2573. Feb 2026




2025
-
 Original Convergent losses of arbuscular mycorrhizal symbiosis in carnivorous plantsNew Phytologist 248: 2040-2051. Sep 2025
Original Convergent losses of arbuscular mycorrhizal symbiosis in carnivorous plantsNew Phytologist 248: 2040-2051. Sep 2025 -
 Original Emergence of isochorismate-based salicylic acid biosynthesis within BrassicalesProceedings of the National Academy of Sciences of the United States of America 122: e2506170122. Jul 2025
Original Emergence of isochorismate-based salicylic acid biosynthesis within BrassicalesProceedings of the National Academy of Sciences of the United States of America 122: e2506170122. Jul 2025 -
 Original Genomic signature and evolutionary history of completely cleistogamous lineages in the non-photosynthetic orchid GastrodiaProceedings of the Royal Society B: Biological Sciences 292: 20250574. May 2025
Original Genomic signature and evolutionary history of completely cleistogamous lineages in the non-photosynthetic orchid GastrodiaProceedings of the Royal Society B: Biological Sciences 292: 20250574. May 2025 - Original Convergent acquisition of disulfide-forming enzymes in malodorous flowersScience 388: 656-661. May 2025
- Review Construction costs and tradeoffs in carnivorous pitcher plant leaves: towards a pitcher leaf economic spectrumAnnals of Botany 135: 1261-1280. Jun 2025

2024
- Original Convergent emergence of glucomannan β-galactosyltransferase activity in asterids and rosidsPlant & Cell Physiology 65: 2030–2039. Oct 2024
2023
-
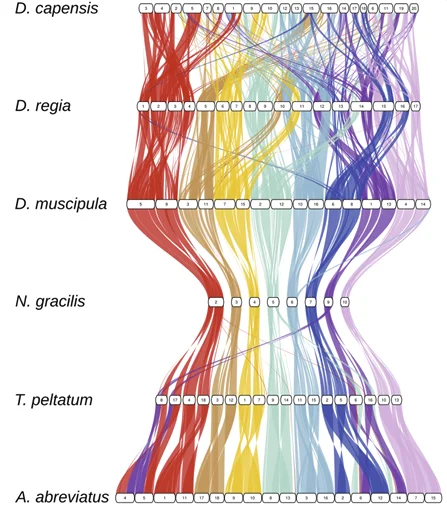
 Original Subgenome dominance shapes novel gene evolution in the decaploid pitcher plant Nepenthes gracilisNature Plants 9: 2000–2015. Dec 2023
Original Subgenome dominance shapes novel gene evolution in the decaploid pitcher plant Nepenthes gracilisNature Plants 9: 2000–2015. Dec 2023 -
 Dispatch Carnivorous plants: Unlocking the secrets of peristome geometry in pitcher plantsCurrent Biology 33: R1155–R1157. Nov 2023
Dispatch Carnivorous plants: Unlocking the secrets of peristome geometry in pitcher plantsCurrent Biology 33: R1155–R1157. Nov 2023 -
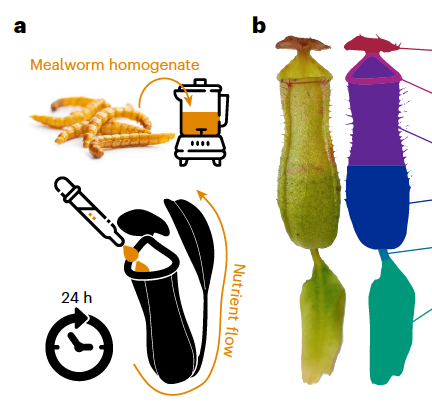
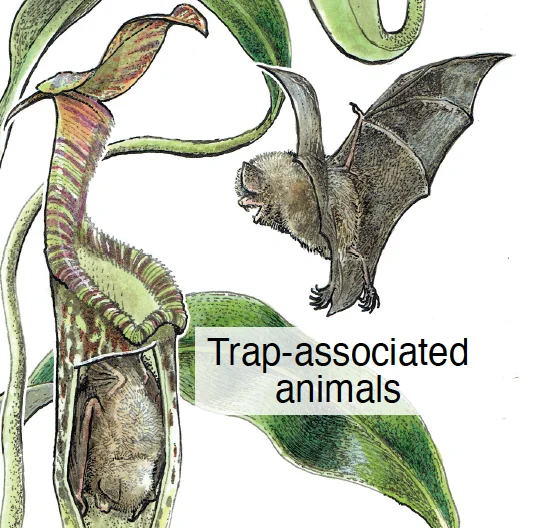 Primer Non-prey biotic interactions in carnivorous plantsCurrent Biology 33: R497–R500. Jun 2023
Primer Non-prey biotic interactions in carnivorous plantsCurrent Biology 33: R497–R500. Jun 2023 -
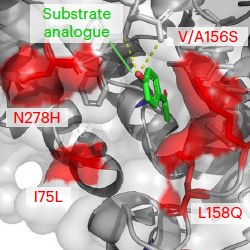 Original Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergenceNature Ecology & Evolution 7: 155–170. Jan 2023
Original Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergenceNature Ecology & Evolution 7: 155–170. Jan 2023 -
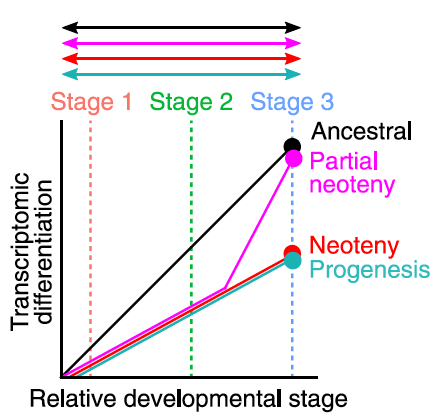 Original Transcriptomic heterochrony and completely cleistogamous flower development in the mycoheterotrophic orchid GastrodiaNew Phytologist 237: 323–338. Jan 2023
Original Transcriptomic heterochrony and completely cleistogamous flower development in the mycoheterotrophic orchid GastrodiaNew Phytologist 237: 323–338. Jan 2023




2022
-
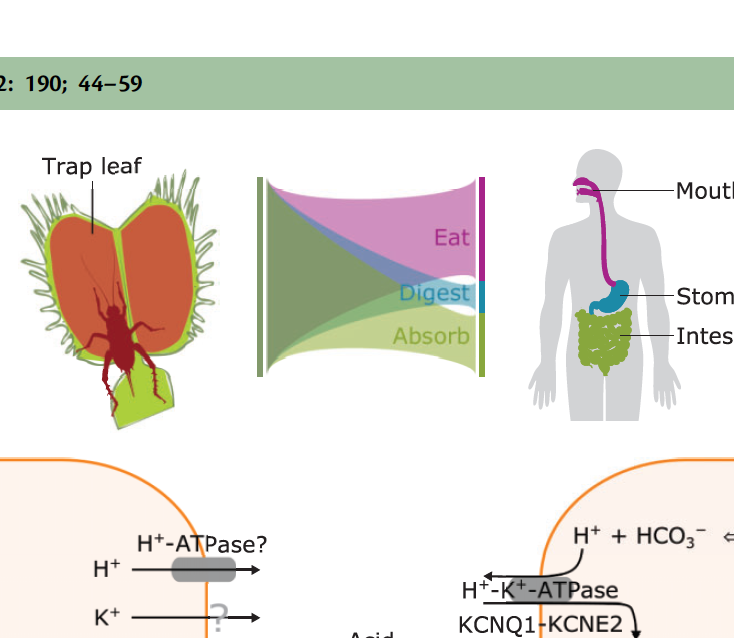 Review The digestive systems of carnivorous plantsPlant Physiology 190: 44–59. Sep 2022
Review The digestive systems of carnivorous plantsPlant Physiology 190: 44–59. Sep 2022 - Preprint Genome sequence of 12 Vigna species as a knowledge base of stress tolerance and resistancebioRxiv : 2022.03.28.486085. Mar 2022

2021
-
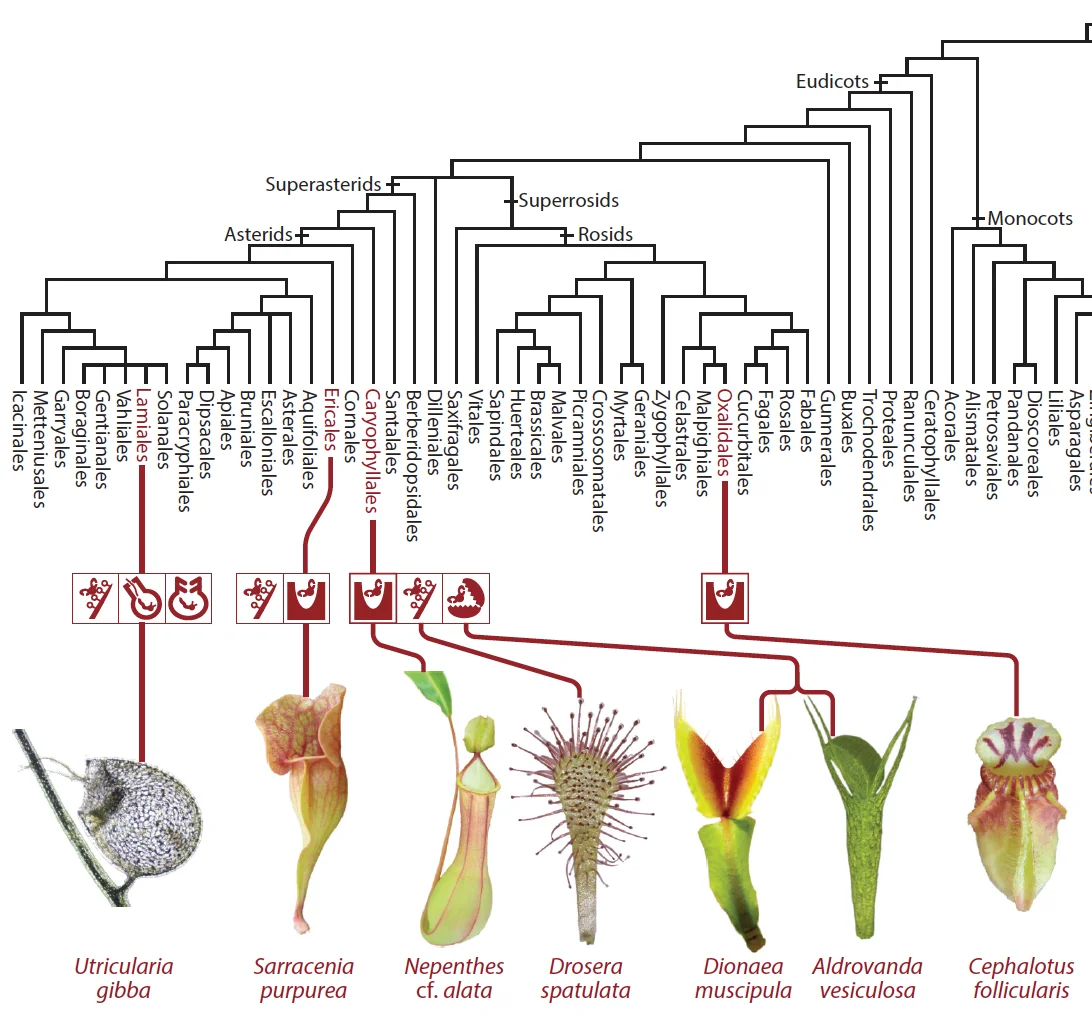 Review On the origin of carnivory: Molecular physiology and evolution of plants on an animal dietAnnual Review of Plant Biology 72: 133–153. Jun 2021
Review On the origin of carnivory: Molecular physiology and evolution of plants on an animal dietAnnual Review of Plant Biology 72: 133–153. Jun 2021 - Original Gene expression evolution in pattern-triggered immunity within Arabidopsis thaliana and across Brassicaceae speciesThe Plant Cell 33: 1863–1887. Jun 2021
-
 Original A discordance of seasonally covarying cues uncovers misregulated phenotypes in the heterophyllous pitcher plant Cephalotus follicularisProceedings of the Royal Society B: Biological Sciences 288: 20202568. Jan 2021
Original A discordance of seasonally covarying cues uncovers misregulated phenotypes in the heterophyllous pitcher plant Cephalotus follicularisProceedings of the Royal Society B: Biological Sciences 288: 20202568. Jan 2021 - Opinion How to grow a tree: Plant voltage-dependent cation channels in the spotlight of evolutionTrends in Plant Science 26: 41–52. Jan 2021

2020
- Original Calcium dynamics during trap closure visualized in transgenic Venus flytrapNature Plants 6: 1219–1224. Oct 2020
-
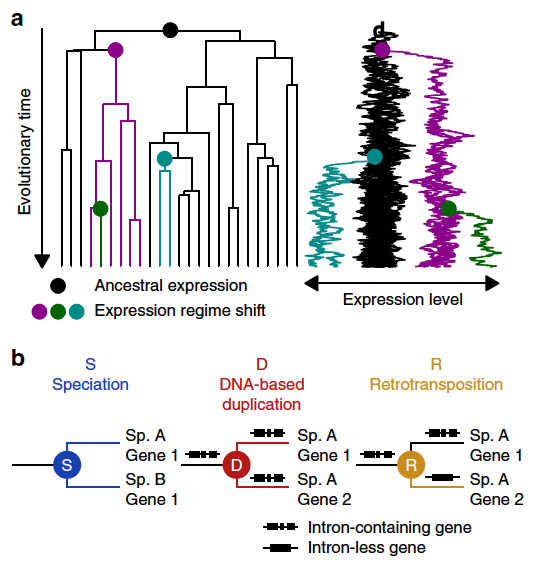 Original Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolutionNature Communications 11: 4459. Sep 2020
Original Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolutionNature Communications 11: 4459. Sep 2020 - Original Genomes of the Venus flytrap and close relatives unveil the roots of plant carnivoryCurrent Biology 30: 2312–2320.e5. May 2020

2019
- Original Assembly and annotation of a draft genome of the medicinal plant Polygonum cuspidatumFrontiers in Plant Science 10: 1274. Oct 2019
2018
- Book Chapter Carnivorous plant genomesCarnivorous plants: Physiology, ecology, and evolution : 135–152. Dec 2018Crossref 11 Dimensions 0
- Preprint Markov katana: A novel method for Bayesian resampling of parameter space applied to phylogenetic treesbioRxiv : 250951. Jan 2018
2017
-
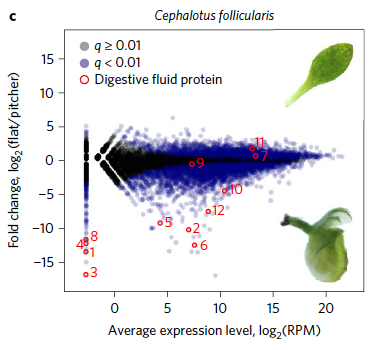 Original Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivoryNature Ecology & Evolution 1: 0059. Feb 2017
Original Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivoryNature Ecology & Evolution 1: 0059. Feb 2017

2015
-
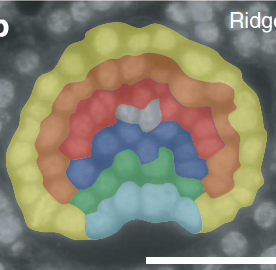 Original Oriented cell division shapes carnivorous pitcher leaves of Sarracenia purpureaNature Communications 6: 6450. Dec 2015
Original Oriented cell division shapes carnivorous pitcher leaves of Sarracenia purpureaNature Communications 6: 6450. Dec 2015

2014
-
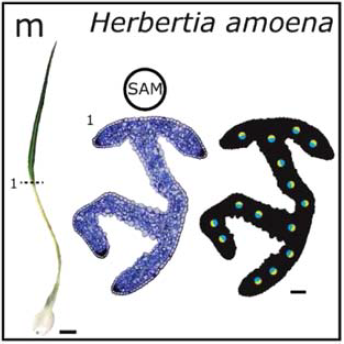 Review Adaxial–abaxial polarity: The developmental basis of leaf shape diversityGenesis 52: 1–18. Jan 2014
Review Adaxial–abaxial polarity: The developmental basis of leaf shape diversityGenesis 52: 1–18. Jan 2014 - Original Molecular phylogeny determined using chloroplast DNA inferred a new phylogenetic relationship of Rorippa aquatica (Eaton) EJ Palmer & Steyermark (Brassicaceae)—lake cressAmerican Journal of Plant Sciences 5: 48–54. Jan 2014Crossref 12 Dimensions 14

2011
-
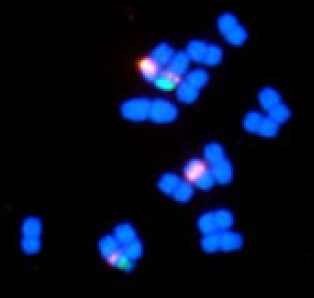 Original Contrasting patterns of the 5S and 45S rDNA evolutions in the Byblis liniflora complex (Byblidaceae)Journal of Plant Research 124: 231–244. Mar 2011
Original Contrasting patterns of the 5S and 45S rDNA evolutions in the Byblis liniflora complex (Byblidaceae)Journal of Plant Research 124: 231–244. Mar 2011

2009
-
 Original Drosera rotundifolia and Drosera tokaiensis suppress the activation of HMC-1 human mast cellsJournal of Ethnopharmacology 125: 90–96. Aug 2009
Original Drosera rotundifolia and Drosera tokaiensis suppress the activation of HMC-1 human mast cellsJournal of Ethnopharmacology 125: 90–96. Aug 2009

2008
-
 Original Somatic chromosome differentiation in three species of the Byblis liniflora complex (Byblidaceae)Chromosome Botany 3: 95–99. Dec 2008Crossref 3 Dimensions 3
Original Somatic chromosome differentiation in three species of the Byblis liniflora complex (Byblidaceae)Chromosome Botany 3: 95–99. Dec 2008Crossref 3 Dimensions 3 - Original Tandem repeat rDNA sequences derived from parents were stably maintained in hexaploids of Drosera spathulata complex (Droseraceae)Cytologia 73: 313–325. Sep 2008Crossref 18 Dimensions 21
- Original Bitter gourd suppresses lipopolysaccharide-induced inflammatory responsesJournal of Agricultural and Food Chemistry 56: 4004–4011. Jun 2008Crossref 75 Dimensions 80
2007
- Original A comparative study of karyotypes in two species of Byblis (Byblidaceae)Chromosome Botany 2: 39–43. Jun 2007Crossref 3 Dimensions 3