publications
Publications from the Fukushima Lab. Authors who contributed as members of the Fukushima Lab are shown in bold. Preview images are attached to papers in which lab members are (co-)first or (co-)corresponding authors. †: co-first authors. *: (co-)corresponding authors.
2026
-
 Species Spotlight Australian pitcher plant (Cephalotus follicularis)Nature Ecology & Evolution : . Feb 2026
Species Spotlight Australian pitcher plant (Cephalotus follicularis)Nature Ecology & Evolution : . Feb 2026 -
 News & Views Genomic cradle for thousandsNature Plants 12: 271-272. Jan 2026
News & Views Genomic cradle for thousandsNature Plants 12: 271-272. Jan 2026 -
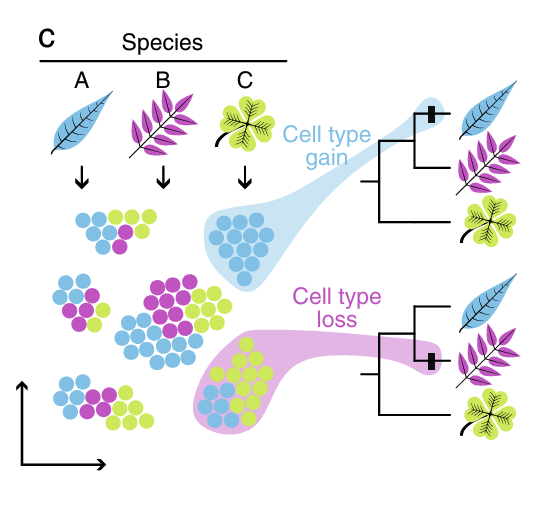 Review Decoding plant cell heterogeneity and dynamics across responses, development, to evolution with single-cell technologiesCurrent Opinion in Plant Biology 90: 102854. Apr 2026
Review Decoding plant cell heterogeneity and dynamics across responses, development, to evolution with single-cell technologiesCurrent Opinion in Plant Biology 90: 102854. Apr 2026 -
 Original Transcriptomic prey-capture responses in convergently evolved carnivorous pitcher plantsNew Phytologist 249: 2559-2573. Feb 2026
Original Transcriptomic prey-capture responses in convergently evolved carnivorous pitcher plantsNew Phytologist 249: 2559-2573. Feb 2026




2025
-
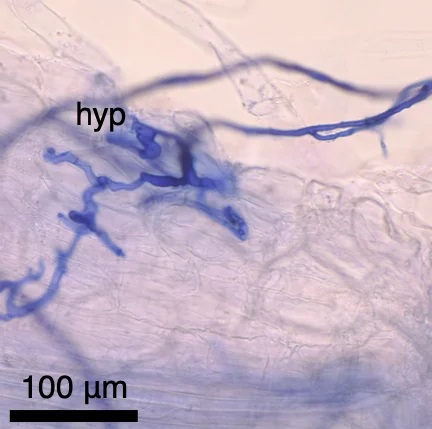 Original Convergent losses of arbuscular mycorrhizal symbiosis in carnivorous plantsNew Phytologist 248: 2040-2051. Sep 2025
Original Convergent losses of arbuscular mycorrhizal symbiosis in carnivorous plantsNew Phytologist 248: 2040-2051. Sep 2025 -
 Original Emergence of isochorismate-based salicylic acid biosynthesis within BrassicalesProceedings of the National Academy of Sciences of the United States of America 122: e2506170122. Jul 2025
Original Emergence of isochorismate-based salicylic acid biosynthesis within BrassicalesProceedings of the National Academy of Sciences of the United States of America 122: e2506170122. Jul 2025 -
 Original Genomic signature and evolutionary history of completely cleistogamous lineages in the non-photosynthetic orchid GastrodiaProceedings of the Royal Society B: Biological Sciences 292: 20250574. May 2025
Original Genomic signature and evolutionary history of completely cleistogamous lineages in the non-photosynthetic orchid GastrodiaProceedings of the Royal Society B: Biological Sciences 292: 20250574. May 2025 - Original Convergent acquisition of disulfide-forming enzymes in malodorous flowersScience 388: 656-661. May 2025
- Review Construction costs and tradeoffs in carnivorous pitcher plant leaves: towards a pitcher leaf economic spectrumAnnals of Botany 135: 1261-1280. Jun 2025

2024
- Original Convergent emergence of glucomannan β-galactosyltransferase activity in asterids and rosidsPlant & Cell Physiology 65: 2030–2039. Oct 2024
2023
-
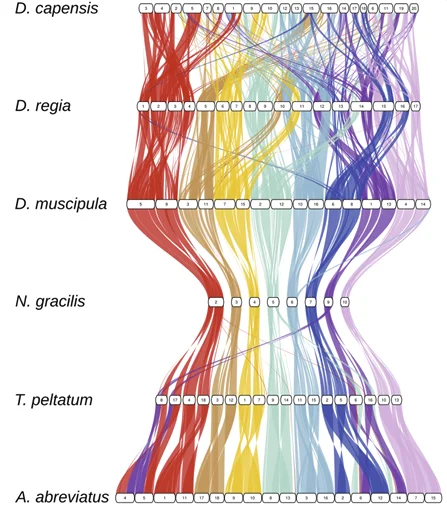
 Original Subgenome dominance shapes novel gene evolution in the decaploid pitcher plant Nepenthes gracilisNature Plants 9: 2000–2015. Dec 2023
Original Subgenome dominance shapes novel gene evolution in the decaploid pitcher plant Nepenthes gracilisNature Plants 9: 2000–2015. Dec 2023 -
 Dispatch Carnivorous plants: Unlocking the secrets of peristome geometry in pitcher plantsCurrent Biology 33: R1155–R1157. Nov 2023
Dispatch Carnivorous plants: Unlocking the secrets of peristome geometry in pitcher plantsCurrent Biology 33: R1155–R1157. Nov 2023 -
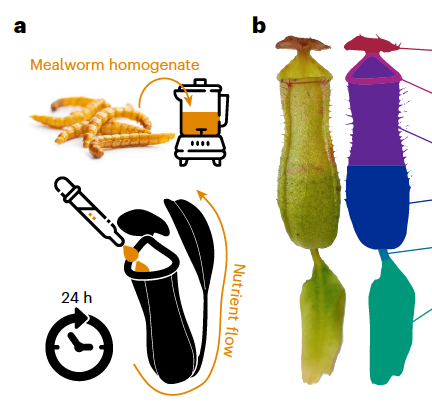
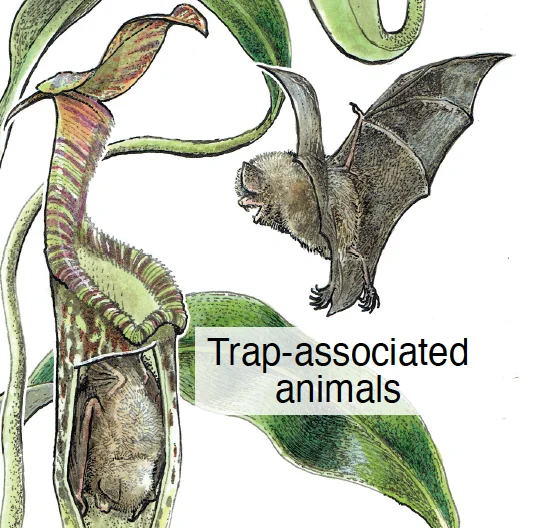 Primer Non-prey biotic interactions in carnivorous plantsCurrent Biology 33: R497–R500. Jun 2023
Primer Non-prey biotic interactions in carnivorous plantsCurrent Biology 33: R497–R500. Jun 2023 -
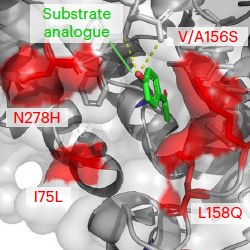 Original Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergenceNature Ecology & Evolution 7: 155–170. Jan 2023
Original Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergenceNature Ecology & Evolution 7: 155–170. Jan 2023 -
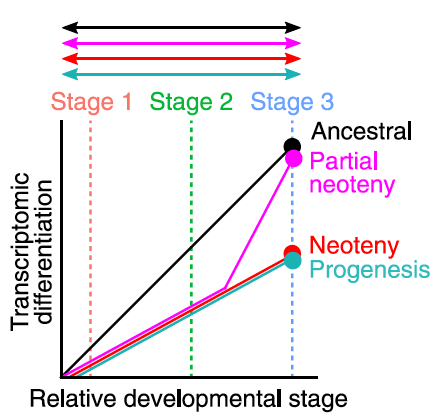 Original Transcriptomic heterochrony and completely cleistogamous flower development in the mycoheterotrophic orchid GastrodiaNew Phytologist 237: 323–338. Jan 2023
Original Transcriptomic heterochrony and completely cleistogamous flower development in the mycoheterotrophic orchid GastrodiaNew Phytologist 237: 323–338. Jan 2023




2022
-
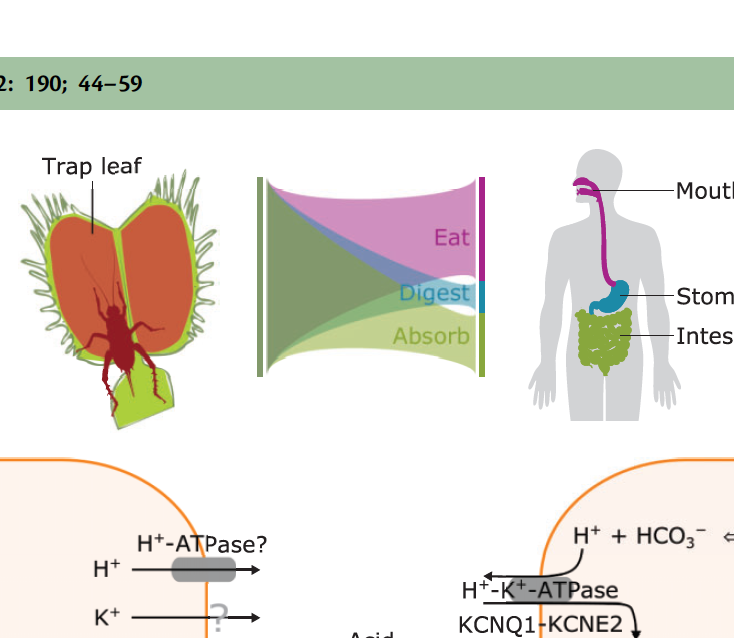 Review The digestive systems of carnivorous plantsPlant Physiology 190: 44–59. Sep 2022
Review The digestive systems of carnivorous plantsPlant Physiology 190: 44–59. Sep 2022 - Preprint Genome sequence of 12 Vigna species as a knowledge base of stress tolerance and resistancebioRxiv : 2022.03.28.486085. Mar 2022

2021
-
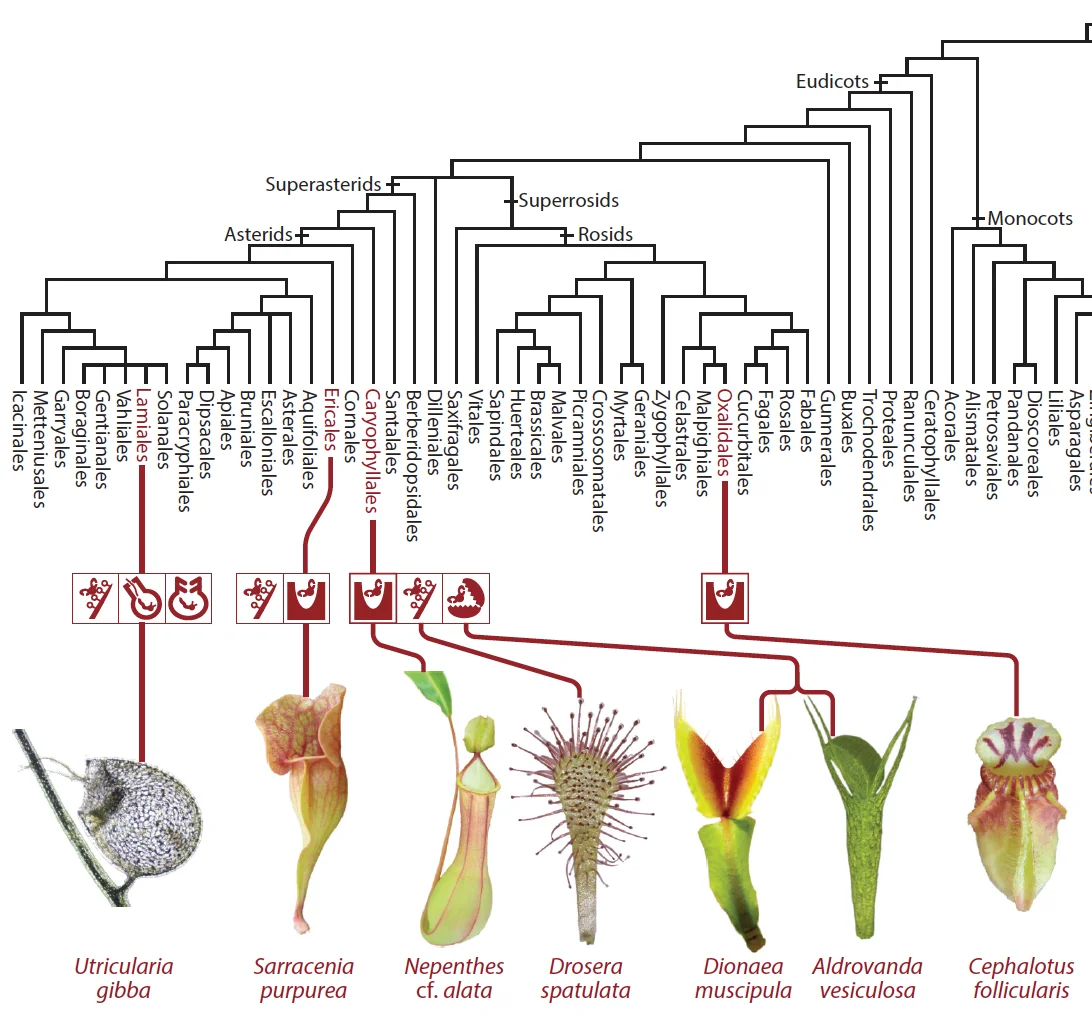 Review On the origin of carnivory: Molecular physiology and evolution of plants on an animal dietAnnual Review of Plant Biology 72: 133–153. Jun 2021
Review On the origin of carnivory: Molecular physiology and evolution of plants on an animal dietAnnual Review of Plant Biology 72: 133–153. Jun 2021 - Original Gene expression evolution in pattern-triggered immunity within Arabidopsis thaliana and across Brassicaceae speciesThe Plant Cell 33: 1863–1887. Jun 2021
-
 Original A discordance of seasonally covarying cues uncovers misregulated phenotypes in the heterophyllous pitcher plant Cephalotus follicularisProceedings of the Royal Society B: Biological Sciences 288: 20202568. Jan 2021
Original A discordance of seasonally covarying cues uncovers misregulated phenotypes in the heterophyllous pitcher plant Cephalotus follicularisProceedings of the Royal Society B: Biological Sciences 288: 20202568. Jan 2021 - Opinion How to grow a tree: Plant voltage-dependent cation channels in the spotlight of evolutionTrends in Plant Science 26: 41–52. Jan 2021

2020
- Original Calcium dynamics during trap closure visualized in transgenic Venus flytrapNature Plants 6: 1219–1224. Oct 2020
-
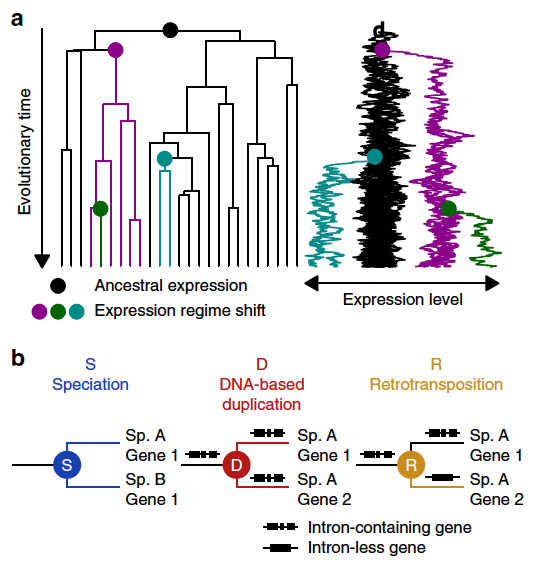 Original Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolutionNature Communications 11: 4459. Sep 2020
Original Amalgamated cross-species transcriptomes reveal organ-specific propensity in gene expression evolutionNature Communications 11: 4459. Sep 2020 - Original Genomes of the Venus flytrap and close relatives unveil the roots of plant carnivoryCurrent Biology 30: 2312–2320.e5. May 2020

2019
- Original Assembly and annotation of a draft genome of the medicinal plant Polygonum cuspidatumFrontiers in Plant Science 10: 1274. Oct 2019
2018
- Book Chapter Carnivorous plant genomesCarnivorous plants: Physiology, ecology, and evolution : 135–152. Dec 2018Crossref 11 Dimensions 0
- Preprint Markov katana: A novel method for Bayesian resampling of parameter space applied to phylogenetic treesbioRxiv : 250951. Jan 2018
2017
-
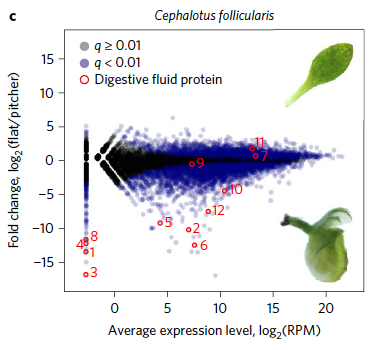 Original Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivoryNature Ecology & Evolution 1: 0059. Feb 2017
Original Genome of the pitcher plant Cephalotus reveals genetic changes associated with carnivoryNature Ecology & Evolution 1: 0059. Feb 2017

2015
-
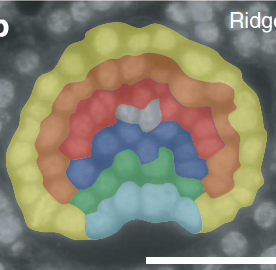 Original Oriented cell division shapes carnivorous pitcher leaves of Sarracenia purpureaNature Communications 6: 6450. Dec 2015
Original Oriented cell division shapes carnivorous pitcher leaves of Sarracenia purpureaNature Communications 6: 6450. Dec 2015

2014
-
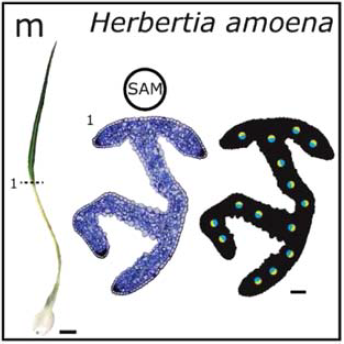 Review Adaxial–abaxial polarity: The developmental basis of leaf shape diversityGenesis 52: 1–18. Jan 2014
Review Adaxial–abaxial polarity: The developmental basis of leaf shape diversityGenesis 52: 1–18. Jan 2014 - Original Molecular phylogeny determined using chloroplast DNA inferred a new phylogenetic relationship of Rorippa aquatica (Eaton) EJ Palmer & Steyermark (Brassicaceae)—lake cressAmerican Journal of Plant Sciences 5: 48–54. Jan 2014Crossref 12 Dimensions 14

2011
-
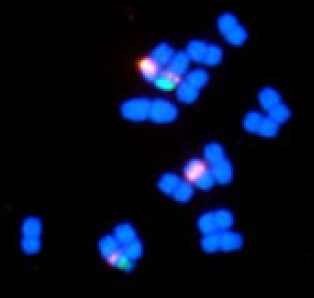 Original Contrasting patterns of the 5S and 45S rDNA evolutions in the Byblis liniflora complex (Byblidaceae)Journal of Plant Research 124: 231–244. Mar 2011
Original Contrasting patterns of the 5S and 45S rDNA evolutions in the Byblis liniflora complex (Byblidaceae)Journal of Plant Research 124: 231–244. Mar 2011

2009
-
 Original Drosera rotundifolia and Drosera tokaiensis suppress the activation of HMC-1 human mast cellsJournal of Ethnopharmacology 125: 90–96. Aug 2009
Original Drosera rotundifolia and Drosera tokaiensis suppress the activation of HMC-1 human mast cellsJournal of Ethnopharmacology 125: 90–96. Aug 2009

2008
-
 Original Somatic chromosome differentiation in three species of the Byblis liniflora complex (Byblidaceae)Chromosome Botany 3: 95–99. Dec 2008Crossref 3 Dimensions 3
Original Somatic chromosome differentiation in three species of the Byblis liniflora complex (Byblidaceae)Chromosome Botany 3: 95–99. Dec 2008Crossref 3 Dimensions 3 - Original Tandem repeat rDNA sequences derived from parents were stably maintained in hexaploids of Drosera spathulata complex (Droseraceae)Cytologia 73: 313–325. Sep 2008Crossref 18 Dimensions 21
- Original Bitter gourd suppresses lipopolysaccharide-induced inflammatory responsesJournal of Agricultural and Food Chemistry 56: 4004–4011. Jun 2008Crossref 75 Dimensions 80
2007
- Original A comparative study of karyotypes in two species of Byblis (Byblidaceae)Chromosome Botany 2: 39–43. Jun 2007Crossref 3 Dimensions 3